Using gnomAD to Drive Insights in Precision Medicine

Originally launched in 2014, the Genome Aggregation Database (gnomAD) is a collaborative effort to collect data from large-scale exome and genome sequencing projects and make them publicly available for scientific use. The Broad Institute-based team recently announced a major update to the database (v4.0), adding data from more than 700,000 individuals and increasing the diversity of represented populations.

Why do datasets like gnomAD matter?
The human genome exhibits on average 4 to 5 million variants from person to person, but most of these variations are not associated with disease. Amidst this range of natural variability, how do researchers hone in on specific variations that are potentially linked to disease? That’s where reference datasets come in – collections of genomic data from many individuals that can be systematically analyzed for patterns associated with disease phenotypes. However, in order to be useful for comparative analyses, reference datasets need to be prepared and harmonized to account for differences in data collection and other confounding factors. gnomAD provides just such a resource for researchers: an extensive, rigorously harmonized set of genomic and exomic data.
How does gnomAD help drug discovery and biomedical research?
Identifying and studying disease-linked genetic variants is crucial for targeted drug discovery and development. In a 2020 Nature study, Dr. Nicola Whiffin et al. leveraged gnomAD data to investigate the impact of natural variation in the LRRK2 gene, which is implicated in Parkinson’s disease. Gain-of-function mutations in this gene are known to increase the risk of Parkinson’s, making it a potential druggable target. However, the consequences of its inhibition are not well understood, and in fact have shown toxicity in animal models. By looking at individuals with natural loss-of-function LRRK2 mutations, the authors found that these mutations were not associated with the adverse consequences shown in animal studies.
Since loss-of-function (LoF) mutations offer a relatively simplified opportunity to study the relationship between gene and disease phenotype, they are often used to demonstrate the effects of targeted drug intervention. Dr. Erik Minikel et al. used gnomAD data as a lens to better understand, and prescribe improvements for, therapeutic studies aimed at these mutations. They conclude that studying isolated LoF mutations has limited application to humans in vivo, as genes work within a complex genomic context.
This context includes regions that don’t necessarily code for proteins. In another Nature 2020 article using gnomAD data, Dr. Whiffin and colleagues demonstrate the impact of untranslated regions on disease phenotypes. Drs. Whiffin and Minikel’s work both show that rigorous analysis of whole-genome data is necessary to evaluate drug targets and more accurately predict their effects.
Why is the latest update important?
First, the sheer volume of data available in the latest update – 5X more than the previous two iterations combined – helps researchers better characterize variations of all frequencies, including rare ones that may not show up in smaller datasets. Every additional genome helps the overall database inch closer to approximating the natural variability of the human population.
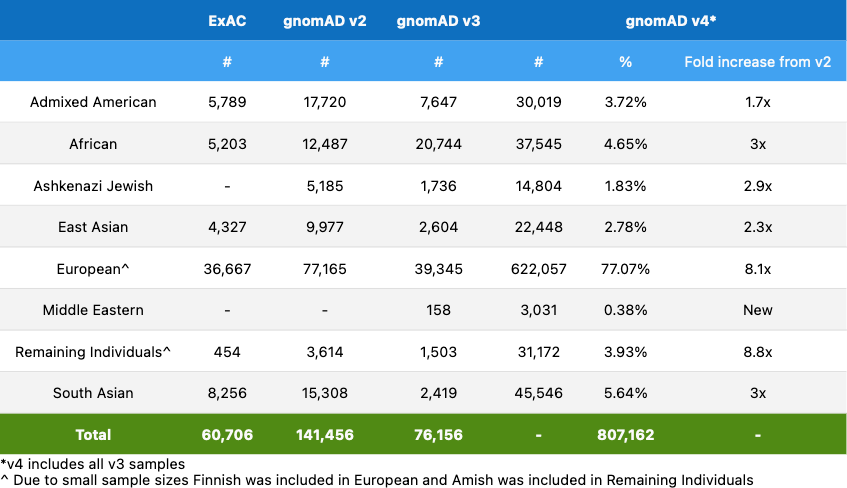
Second, while a large proportion of the data is from individuals with European ancestry, v4 does exhibit a 2-3 fold increase in representation of individuals from non-European backgrounds. As many drug developers use these kinds of datasets to discover and validate therapeutic targets, as well as segment potential patient populations, lack of representation can be a matter of life or death for underrepresented groups.
Third, the update addresses inconsistencies in standards that are endemic to bioinformatics. Specifically, the team standardized naming conventions for certain fields like genetic ancestry subgroups. Standardization allows researchers to more easily compare different datasets and conduct multi-omic analyses.
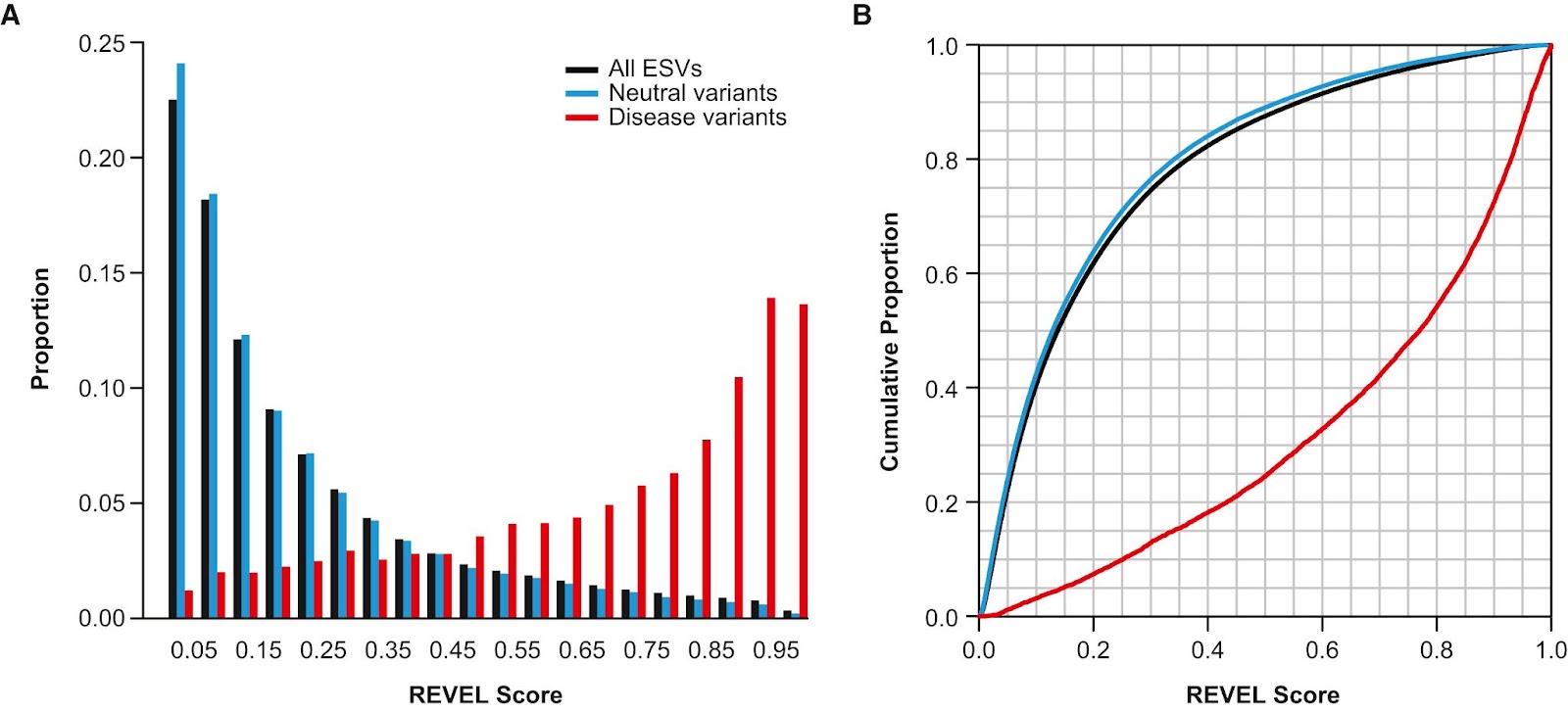
Lastly, v4 helps researchers grasp the predicted functional importance of a particular variant with updated annotations, calculated from 5 different resources (REVEL, CADD, phyloP, spliceAI, and Pangolin). Using multiple methods for functional prediction helps improve confidence in the actual effect of a variant.
How can I use gnomAD in my research?
While public datasets like gnomAD are rich in information, tackling them without the proper tools can waste a lot of time and resources. Here are several key points to keep in mind when preparing to work with a large public dataset:
1. You will need adequate infrastructure to store, manage, and eventually run analyses on the data. When working with this amount of data, sparse or slow compute resources can render your project infeasible. Make sure you have the computational power you need to efficiently run your analyses.
2. The data also need to be curated to serve your particular analysis goals. These steps take time, but it’s worth the investment to do them correctly at the beginning of the project.
3. Without consistent management and tracking provenance practices, your results can become inscrutable to collaborators or reviewers. Ensure your data uphold FAIR practices: they should be Findable, Accessible, Interoperable, and Reusable.
4. Even if you have computational experience, setting up and troubleshooting analysis pipelines can quickly become a full-time job without access to engineering support.
5. Accurately interpreting results is key to generating actionable insights. Bioinformatics expertise is invaluable to every impactful data-driven study.
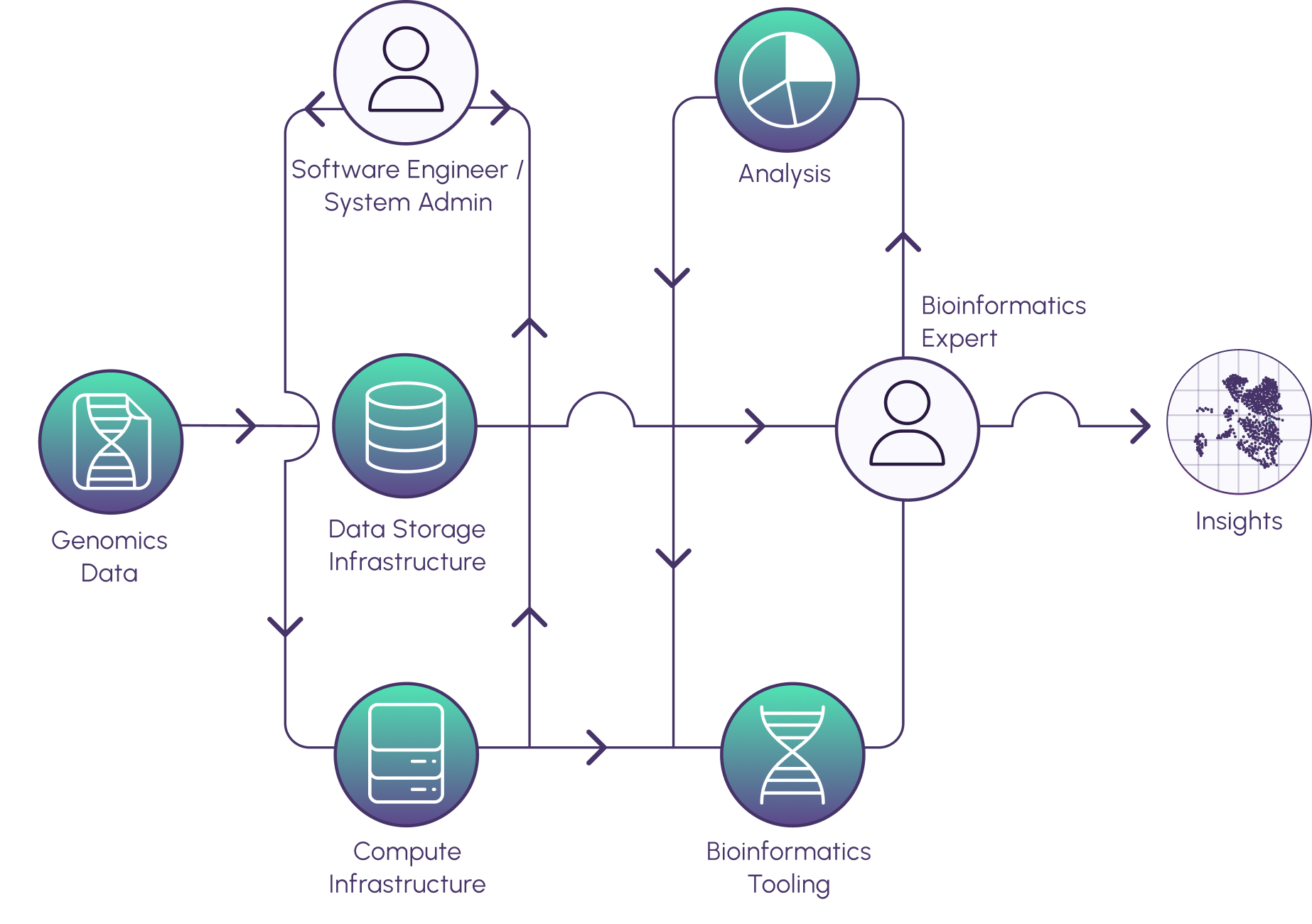
Watershed offers a streamlined solution to all of these problems and more. We empower researchers with the tools they need to extract meaningful insights and make breakthrough discoveries using gnomAD and other public datasets every day. Get in touch with us at contact@watershed.bio or schedule a demo today!